
Экстрапирамидная система является сложным комплексом, состоящим из ряда тесно соединенных между собой ядер серого вещества. С функциональной точки зрения, данный комплекс представляет собой базу реализации сложных безусловных рефлексов (врожденных, видовых), называемых инстинктами (оборонительный, пищевой, половой, материнский и другие).
Функцией экстрапирамидного комплекса является также регуляция мышечного тонуса, нормальных содружественных движений (синкинезии) и рефлекторных двигательных реакций: содружественные движения при ходьбе, жестикуляция, отдергивание руки от горячего предмета или при болезненном воздействии. Еще одной функцией данной системы является обеспечение постуральных рефлексов (позы, положение в пространстве). Лечение экстрапирамидных расстройств, как правило, длится всю жизнь.
Экстрапирамидная система
Основные образования, из которых состоит экстрапирамидная система:
- чечевицеобразное ядро;
- хвостатое ядро;
- красное ядро;
- черная субстанция;
- субталамическое ядро;
- ретикулярная формация;
- ядра мозжечка;
- премоторная кора (эта зона имеет непосредственное отношение как к экстрапирамидной системе, так и к пирамидной).
Данные анатомические образования имеют тесные связи и между собой и с другими образованиями центральной нервной системы.
Именно нарушениями в работе клеток, составляющих образования экстрапирамидной системы, а также их проводящих путей, обусловлено возникновение экстрапирамидных расстройств. Данная группа заболеваний характеризуется многообразием симптоматики.
Какие бывают экстрапирамидные расстройства
Современная классификация экстрапирамидных расстройств подразделяет их на две большие группы:
- 1. гипокинетические (паркинсонизм, изолированная акинезия);
- 2. гиперкинетические.
Гиперкинетические расстройства по своему характеру возникновения могут быть:
- спонтанными (хорея, баллизм, разновидности миоклонии);
- акционными или кинезиогенными, которые провоцируются произвольными движениями (некоторые разновидности пароксизмальных дискинезий, кинетический тремор, дистония);
- специфическими кинезиогенными — гиперкинезы, возникающие лишь при выполнении определенных движений (например, спазм при письме или игре на музыкальных инструментах);
- рефлекторными — вызванные раздражениями извне (например, рефлекторная миоклония).
Исходя из двигательного рисунка, гиперкинетические расстройства классифицируют следующим образом:
- ритмические — вызванные периодическим сокращением мышц агонистов и антагонистов (дрожание, миоритмия);
- преимущественно тонические — провоцируемые одновременным сокращением мышц агонистов и антагонистов (дистония);
- преимущественно клонические (быстрые, мобильные, фазические), проявляющиеся как в виде простых движений, вызванных сокращением одной мышцы, так и в виде сложных по организации движений, похожих на нормальный двигательный акт (тики, хорея).
По распространенности гиперкинезы делят на генерализованные, сегментарные, фокальные и мультифокальные.
Отдельные экстрапирамидные нарушения
Клиническая картина и варианты течения расстройств экстрапирамидной системы зависят от зоны и механизма поражения.
Болезнь Паркинсона
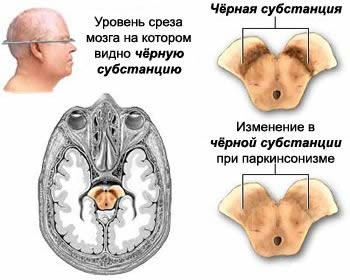
Данную патологию принято относить к группе дегенеративных заболеваний центральной нервной системы. Патогенетическим фактором патологии является гибель нейронов ряда образований экстрапирамидной системы:
- черной субстанции;
- бледного шара;
- скорлупы.
Этиология данных нарушений на сегодня не установлена, но существуют данные о генетической предрасположенности.
Следует различать:
- Болезнь Паркинсона (первичная форма): в структуре всех случаев паркинсонизма это состояние составляет большинство случаев (80%). Возникновение патологии обусловлено генетическими факторами.
- Вторичный паркинсонизм, или синдром Паркинсона. Является одним из симптомов различных поражений или заболеваний центральной нервной системы.
Клиническая картина
Происходит постепенное замедление и появление скованности активных движений: отмечается, что раньше всего изменения становятся заметны в правой руке у правшей, поскольку нарушения вызывают в разной степени выраженные трудности при письме и выполнении мелких действий пальцами. Затем, по мере прогрессирования тремора и скованности, изменения становятся заметны и в других частях тела:
- Появляется гипертонус скелетных мышц, благодаря чему постепенно формируется определенная поза, характеризующаяся сутулостью и сгибанием конечностей.
- Наблюдаются характерные изменения в походке: она становится “шаркающей”, “семенящей”. Появляется пропульсия: такому пациенту, как правило, сложно шагнуть при начале ходьбы, а затем, в результате смещения вперед центра тяжести он не может остановиться. Из-за этого больной теряет равновесие. Постепенно стираются индивидуальные особенности походки.
- Начинает дрожать подбородок и конечности. Особенно это заметно в состоянии покоя.
- Отмечается застывание, бедность мимики, редкое моргание веками — так называемое маскообразное лицо.
- Снижается отчетливость, внятность речи.
- Возможна гиперсаливация.
- В разной степени замедляется темп мышления.
- Происходит снижение внимания при сохранности на первых стадиях заболевания интеллектуальных способностей и памяти.
- Из поведенческих изменений: у части больных примерно в половине случаев пропадает мотивация, сужается круг интересов, снижается аффективный фон, вплоть до хронической депрессии.
- Вегетативные нарушения будут выражаться в расстройствах мочеиспускания, запорах, импотенции снижении обоняния, гиперпродукции кожного жира, ведущей к сальности кожи и волос.
Заболевания, вызывающие вторичный паркинсонизм
Причинами данного расстройства могут стать следующие действия и патологии:
- Атеросклероз церебральных сосудов.
- Употребление пациентом некоторых лекарственных препаратов (например, некоторые нейролептики).
- Интоксикации, в частности, этиловым спиртом, угарным газом, цианистыми соединениями, марганцем.
- Наркомания.
- Другие дегенеративные заболевания ЦНС — они способны затронуть экстрапирамидную сферу и, таким образом, вызвать паркинсонизм.
- Воспалительные процессы, поражающие ЦНС (энцефалиты).
- Онкологическое поражение головного мозга.
- Некоторые наследственные заболевания.
- Черепно-мозговые травмы, в том числе повторные повреждения головы (даже не очень тяжелые).
Чтобы определиться с формой заболевания (первичная или вторичная), следует выявить наличие в анамнезе условий, способствующих возникновению паркинсонизма:
- выраженный атеросклероз;
- гипертоническая болезнь;
- употребление токсических веществ, способных спровоцировать данную патологию;
- перенесенные больным нейроинфекции или черепно-мозговая травма.
Лечение
Применение противопаркинсонических медикаментов целесообразно начинать, когда уже появились двигательные ограничения. Для лечения используются следующие группы препаратов:
- леводопы;
- амантадин;
- ингибиторы моноаминоксидазы;
- синергисты дофамина;
- антихолинергические средства (Циклодол).
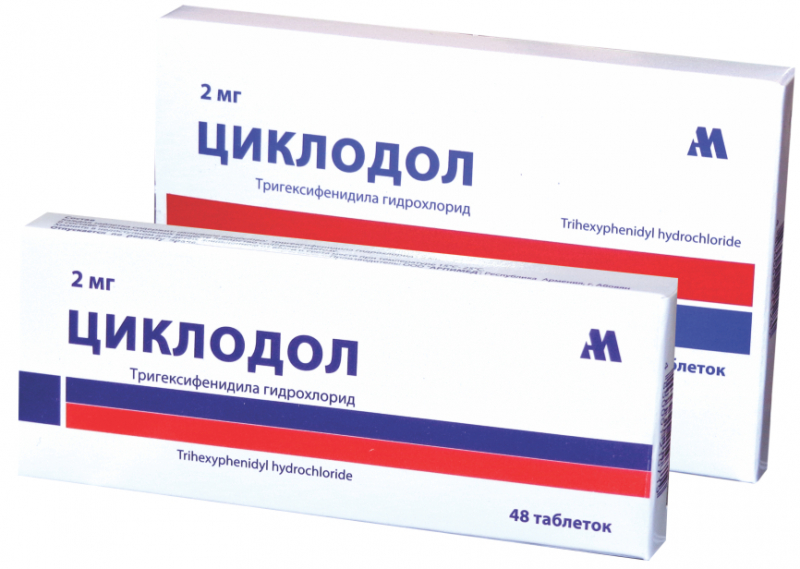
Для достижения стабильного результата дозы лекарственных средств необходимо постоянно повышать. Следует помнить, что данная особенность приведет к неизбежному развитию побочных реакций. Чтобы сгладить это нежелательное явление, следует начинать лечение с легких, низко-дозированных лекарств, а дозировку увеличивать только в случае крайней необходимости. Хороший результат дает комбинирование препаратов, имеющих различные механизмы действия.
Симптоматическая терапия: вегетативные расстройства устраняют при помощи средств, эффективных при запорах; депрессию, повышенную тревожность и диссомнию снимают антидепрессанты. Хорошо сочетаются с медикаментозным лечением народные средства, влияющие на эти же симптомы.
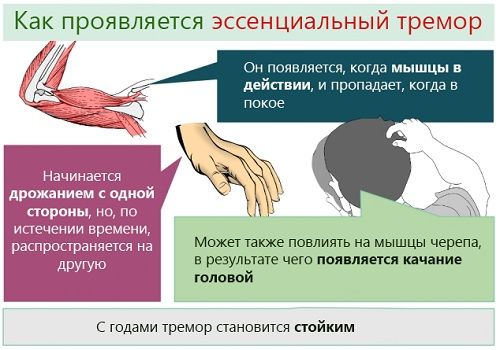
Эссенциальный тремор (доброкачественный, семейный тремор)
Это постуральный тремор, наблюдающийся у здоровых в остальном людей, нередко у нескольких членов одной семьи. Аномалия имеет аутосомно-доминантный механизм наследования. Патогенез и его возникновение на сегодняшний день не установлены.
Заболевание чаще дебютирует у пожилых и лиц среднего возраста.
Характерна локализация тремора в одной или обеих руках и голове. На нижние конечности гиперкинетическое расстройство не распространяется. У некоторых пациентов отмечается дрожание голоса.
Со временем тремор может прогрессировать, но к инвалидности не приводит, за исключением тех случаев, когда работа человека связана с письмом или другими движениями, которые задействуют мелкую моторику рук.
На фоне приема небольших доз алкоголя тремор несколько снижается.
Лечение
Медикаментозную терапию рекомендуется назначать как можно позже, т. к. она должна продолжаться пожизненно. Хорошую эффективность показал Пропранолол в дозе от 40 до 120 мг дважды в сутки.
Такую же дозировку, но однократно рекомендуется принимать перед воздействием провоцирующих факторов.
Если тремор выражен несущественно, лечение не требуется.
Прогрессирующий надъядерный паралич (синдром Стила-Ричардсона-Ольшевского)
Это редкое заболевание дегенеративной природы, имеющее спорадический характер.
Основой патогенеза является дегенерация и гибель нейронов среднего мозга, варолиева моста, базальных ядер и зубчатого ядра мозжечка. Характерно и снижение содержания дофамина и гамма-линоленовой кислоты хвостатого ядра и скорлупы.
Заболевание дебютирует в возрасте от 45 до 75 лет. Чаще болеют мужчины (встречается в 2 раза чаще, чем у женщин).
Характерным признаком является надъядерная офтальмоплегия. С нее и начинается заболевание, а позже присоединяется горизонтальный парез взора.
Для поздних стадий характерно нарушение плавных следящих движений глаз.
Нередко присоединяется мышечная дистония, в основном разгибателей шеи, ригидность мышц конечностей и гипокинезия. После проявления таких отклонений необходим дифференциальный диагноз болезни Паркинсона и симптоматического паркинсонизма.
Специфическими признаками, отличающими прогрессирующий надъядерный паралич от болезни Паркинсона, являются:
- выраженный парез взора вниз;
- горизонтальный парез взора;
- преобладание тонуса разгибателей туловища;
- отсутствие тремора.
В случае начала фармакотерапии противопаркинсонические средства при данном заболевании будут неэффективны.
Сочетание офтальмоплегии с ригидностью мышц шеи и туловища провоцирует частое падение.
В некоторых случаях присоединяется нарушение со стороны лицевого нерва, дизартрия, дисфагия. Для продвинутых стадий характерны проявления псевдобульбарного паралича:
- повышение рвотного рефлекса;
- повышение нижнечелюстного рефлекса;
- появление насильственного смеха и плача;
- усиление сухожильных рефлексов;
- патологические разгибательные рефлексы.
Нередко наблюдается снижение интеллекта.
Очаговых нарушений функций коры не регистрируется.
Прогрессирующий надъядерный паралич можно заподозрить в случае, когда человек среднего или пожилого возраста предъявляет жалобы на частые падения, а при осмотре регистрируются экстрапирамидные нарушения, дистонии разгибателей шеи или парез взора вниз.
Лечение
Как правило, медикаментозное лечение малоэффективно. Продолжительность жизни пациентов после дебюта заболевания редко превышает 10 лет. Ригидность и гипокинезию удается уменьшить при помощи дофаминергических средств. М-холиноблокаторы и антидепрессанты помогают улучшить речь, походку и устранить насильственный смех и плач. Но заболевание продолжает прогрессировать. Со временем присоединяется аспирация пищи, развивается истощение.
Кортикобазальная дегенерация
Это редко встречающееся заболевание носит спорадический характер.
Дебют патологии происходит в пожилом возрасте и проявляется поражением одной или нескольких конечностей.
Основное патогенетическое звено — гибель нейронов и глиоз коры головного мозга и черной субстанции.
Неврологическое обследование выявляет симптоматику паркинсонизма. Но диагноз синдрома Паркинсона не объясняет всех клинических проявлений.
На первый план выступают явления апраксии. Позже присоединяются другие расстройства корковых функций:
- афазия;
- агнозия;
- сенсорное невнимание;
- легкая деменция.
Дифференциально-диагностическим признаком является неэффективность противопаркинсонических средств.
Заболевание непрерывно прогрессирует, приводя к инвалидизации и летальному исходу.
Стриатонигральная дегенерация
Название заболевания указывает на наиболее страдающие структуры — черную субстанцию, субталамическое и хвостатое ядро, скорлупу, бледный шар.
Возрастная группа, для которой характерны данное заболевание, — от 60 лет. У мужчин и у женщин встречается с одинаковой частотой.
Характерен синдром паркинсонизма, но тремор выражен в меньшей степени. Интеллектуальная сфера не затронута.
Встречаются вегетативные расстройства, и тогда неврологическое исследование выявляет, кроме экстрапирамидных, пирамидные нарушения и отлконения со стороны мозжечка.
Лечению стриатонигральная дегенерация продается плохо. Противопаркинсонические средства неэффективны.
Торсионная дистония
Данным термином принято называть дистонию, не сопровождающуюся другими неврологическими нарушениями при условии отсутствия родовой травмы.
Патогенез данного расстройства и биохимические нарушения, приводящие к его развитию, изучены недостаточно.
Принято считать, что развитие торсионной дистонии обусловлено патологией базальных ядер.
Диагноз ставится путем последовательного исключения других причин.
Среди всех случаев торсионной дистонии зарегистрированы и спорадическая, и наследственная формы заболевания. Для наследственных характерны следующие пути передачи:
- аутосомно-доминантный;
- аутосомно-рецессивный или рецессивный;
- сцепленный с x-хромосомой.
Начавшись в детском возрасте, заболевание поражает в первую очередь мускулатуру ног. Такая ранняя форма имеет менее благоприятный прогноз, по сравнению с дистонией позднего начала.
Примерно в 30% случаев пациенты в результате патологии теряют способность ходить.

Признаки патологии
Клиническая картина характеризуется насильственными движениями, необычными позами. Начавшееся с ног заболевание распространяется на мышцы шеи, туловища, конечностей и лица. Сначала насильственные движения возникают при попытке произвольных движений. По мере прогрессирования заболевания они становятся постоянными и приводят к инвалидности.
Частным случаем торсионной дистонии является болезнь Сегавы (ДОФА- чувствительная дистония), наследуется по аутосомно-доминантному типу. Начинается в детском возрасте. Мышечная дистония при данной патологии сочетается с гипокинезией и ригидностью.
Лечение
Лечение проводится симптоматическое, но эффективность нередко низкая.
Наиболее действенны м-холиноблокаторы в высоких дозировках. Начинают лечение с низких доз, которые постепенно повышают до получения результата или развития побочных действий.
В некоторых случаях помогает Галоперидол и фенотиазины, но для них характерны побочные эффекты в виде паркинсонизма.
Иногда симптомы удается снизить при помощи Диазепама, Баклофена, Карбамазепина.
При одностороннем поражении показана таламотомия.
При болезни Сегавы высокую эффективность показали препараты леводопы в низких дозах.
Сегментарная дистония
Отличие данного заболевания от торсионной дистонии состоит в том, что насильственные движения в результате нарушения мышечного тонуса регистрируются лишь в отдельных мышечных группах и участках тела.
Существует гипотеза, что данная патология является разновидностью торсионной дистонии.
Блефароспазм, оромандибулярная дистония, спиноцеребеллярная дегенерация могут возникать изолированно.
Блефароспазм — неконтролируемый спазм мышц вокруг глаз.
Оромандибулярная дистония проявляется непроизвольными сокращениями мышц жевательной группы, языка и мимических мышц, расположенных вокруг рта. Внешне она выражается вытягиванием, сморщиванием губ, сжиманием челюстей, движениями и высовыванием языка. В сочетании с блефароспазмом аномалия носит название лицевого спазма Мейджа.
Спастическая кривошея является частным случаем сегментарной дистонии и характеризуется насильственными движениями головы в сторону. Такой гиперкинез может сочетаться с наклонами головы вперед или назад.
В начале заболевания патологический тонус имеет преходящий характер, но по мере прогрессирования нарушение становится постоянным, в результате чего голова у человека все время пребывает в неестественном положении. В первые месяцы характерны спонтанные ремиссии, но это не влияет на прогрессирование патологии.
Лечение
Фармакотерапия в большинстве случаев неэффективна.
Иногда симптоматика несколько регрессирует под воздействием Диазепама, Баклофена, Карбамазепина.

Используются инъекции ботулотоксина типа А в мышцы с нарушением тонуса. Это вещество обладает свойством на время блокировать выделение ацетилхолина из пресинаптической мембраны. Продолжительность эффекта — от нескольких недель до нескольких месяцев. Инъекции препарата можно проводить повторно. На сегодняшний день это наиболее действенный метод лечения большинства разновидностей сегментарной дистонии.
Спиноцеребеллярная дегенерация
Данное заболевание передается по аутосомно-доминантному механизму и дебютирует на третьем или четвертом десятилетии жизни.
Наиболее характерна патология для жителей Португалии.
Клиническая картина характеризуется умеренно выраженными симптомами паркинсонизма. Преобладают спастичность, снижение рефлексов, нарушения со стороны мозжечка, наружная офтальмоплегия. Иногда присоединяется нейропатия.
Интеллектуальная сфера не страдает. Структурные нарушения в головном мозге соответствуют патологоанатомическим отклонениям при стрионигральной дегенерации. Но к поражению черного тела, бледного шара, скорлупы, хвостатого и субталамического ядер присоединяются изменения зубчатого ядра мозжечка.
Лечение заболевания не разработано.
Профессиональный спазм
Это дистоническое нарушение, развивающееся на базе профессиональных навыков и движений, требующих высокой степени точности:
- писчий спазм;
- спазм парикмахера;
- телеграфистки;
- скрипача;
Клинические проявления на примере писчего спазма: насильственное сокращение мускулатуры кисти и предплечья при письме.
По мере прогрессирования патологии дистония мышц появляется и при других действиях — бритье, наложении косметики, приготовлении пищи, пользовании столовыми приборами.
Медикаментозная терапия имеет очень низкую эффективность и пациентам в большинстве случаев приходится учиться пользоваться другой рукой.
Патогенез заболевания находится в стадии изучения. Имеются данные, что возникновение отклонения обусловлено нарушением обработки эфферентных сигналов от пораженной конечности.
Лекарственные экстрапирамидные расстройства
Лекарственные экстрапирамидные расстройства возникают в результате побочных воздействий некоторых лекарственных препаратов:
|
Название расстройства |
Характеристика |
Тактика |
|
Лекарственный паркинсонизм |
Механизмы развития:
Особенности:
Дифференциальная диагностика основана на данных анамнеза |
Отмена нейролептических препаратов и замена их на средства, имеющие более выраженное м-холиноблокирующее действие, или назначение приема м-холиноблокаторов. Прием леводопы неэффективен и способен вызвать психические нарушения |
|
Острые лекарственные гиперкинезы |
Острая дистония, проявляющаяся блефароспазмом, спастической кривошеей и другими гиперкинезами в виде хореи или спазмов лицевой мускулатуры. Развивается в результате длительного лечения блокаторами дофаминовых рецепторов. Симптоматика развивается в течение первой недели лечебного курса, чаще за 1-48 часов. Более характерна для молодого возраста |
Эффективным является применение м-холиноблокаторов (бензатропин, дифенгидрамин) |
|
Поздняя нейролептическая акатизия |
Акатизия — это двигательное беспокойство. Такой пациент испытывает постоянную потребность в движении. Развитие этого эффекта наиболее характерно для длительного применения нейролептиков. Нарушения чаще встречаются у женщин |
Отмена препарата, спровоцировавшего патологию, с заменой на средства, обладающие более выраженным м-холиноблокирующим эффектом |
|
Поздние нейролептические гиперкинезы |
Развиваются в результате продолжительного приема нейролептиков, блокирующих дофаминовые рецепторы. Вероятность развития данного расстройства возрастает с возрастом. Патогенез неизвестен. Имеется гипотеза, что данная патология обусловлена повышением чувствительности дофаминовых рецепторов под действием нейролептиков. Но оно развивается лишь у некоторых больных. Появление гиперкинеза характерно не ранее, чем через полгода после начала терапии. Нарушение проявляется в виде хореоатетоидных движений мышц лица и рта у взрослых и конечностей — у детей. Генерализованное отклонение встречается редко, в основном у пожилых пациентов. Возможно появление тиков. Дифференциальный диагноз — на основании анамнеза |
В молодом возрасте и у детей нарушения часто проходят самостоятельно. У пожилых пациентов — сохраняются на длительный период. Данное отклонение плохо поддается лечению. Может помочь отмена нейролептика. Бывают эффективны м-холиноблокаторы |
|
Злокачественный нейролептический синдром |
Проявления:
Летальность 5-20%. Нарушение развивается у 2-3 % пациентов, проходящих курс лечения нейролептиками. Развитие злокачественного нейролептического синдрома более характерно для молодого возраста. Отклонения могут развиться в любой момент, но чаще — в первый месяц лечения. Обычно симптоматика нарастает в течение 1-2 суток. Патологию следует дифференцировать со следующими состояниями:
От лекарственного паркинсонизма данную патологию отличают наличие вегетативных расстройств и гипертермии. Возможные осложнения:
|
Восстановление занимает 2-3 недели. Резкая отмена препаратов-антипсихотиков вызывает развитие синдрома отмены. Поэтому пациент должен находиться под постоянным наблюдением |
|
Другие двигательные нарушения |
|
Коррекция дозы в сторону ее снижения или отмена спровоцировавшего нарушения препарата |
Синдром Жиль де ла Туретта
Это заболевание неизвестной на сегодняшний день этиологии. Связь происхождения его с этническими или социальными факторами и перинатальной патологией не установлена.
Заболевание дебютирует в возрасте от 5 до 15 лет и протекает с чередующимися обострениями и ремиссиями.
Известны семейные случаи. Тип наследования — аутосомно-доминантный. Чаще болеют мальчики. Патогенез неясен, морфологические изменения не установлены.
Поскольку известно, что симптомы заболевания стираются под воздействием блокаторов дофаминовых рецепторов, считается, что оно обусловлено чрезмерной активностью дофаминергической системы.
Клиническая картина
Характерным проявлением патологии являются множественные хронические двигательные и голосовые тики. Чаще заболевание начинается с двигательных нарушений. Тики чаще вовлекают мышцы лица и проявляются морганием, хмыканьем, подниманием бровей, зажмуриванием, поджиманием губ или подергиванием мимических мышц. Позже присоединяются и другие двигательные тики, затем — голосовые, проявляющиеся в виде мычания, свиста, похрюкивания, вздохов, откашливания, кряхтения. А в ряде случаев отмечено непроизвольное выкрикивание слов, в том числе бранных, непроизвольное повторение услышанных фраз.
Локализация и характер тиков непостоянны, могут меняться. На некоторое время их можно подавить усилием воли. В отдельных случаях встречаются сложные тики: подпрыгивание или повторяющиеся членовредительские действия (кусания ногтей, губ, подергивания за волосы).
Встречаются тики сенсорного характера в виде повторяющихся ощущений: щекотания, холода, тепла, чувства давления.
Тики нередко сопровождаются поведенческими отклонениями: неврозами, навязчивыми состояниями, синдромом нарушения внимания и гиперактивностью.
Среди больных чаще встречаются левши и амбидекстры. Электроэнцефалограмма выявляет неспецифические изменения.
Нередко диагноз устанавливается уже спустя годы после начала заболевания.
Многочисленные тики могут приводить к социальной дезадаптации, вызывающей депрессивное состояние, а в тяжелых случаях и суицидальные намерения.
Дифференциальная диагностика
Дифференциальная диагностика должна проводиться с многочисленными разнообразными тиками, характерными для детского возраста, которые отличаются хорошим прогнозом и не требует терапии. Детские и юношеские тики нередко прекращаются самостоятельно.
Подобные гиперкинезы характерны для болезни Вильсона.
Синдром Гентингтона отличают наличие деменции и особенности гиперкинетических расстройств. Окончательный диагноз ставится с помощью генной диагностики.
Если в анамнезе отсутствует указание на ревматические атаки, полиартрит и поражение сердца, то за синдром Жиль Де Ля Туретта можно принять хорею сиденгама. Но при этом заболевании гиперкинезы исчезают через 3-6 месяцев.
Множественные тики следует дифференцировать с последствиями энцефалита и приема психостимуляторов и нейролептиков.
Лечение
Лечение продолжительное, симптоматической направленности.
У половины больных эффективен Клонидин. При назначении следует учитывать его побочные эффекты: сонливость, слюнотечение, сухость во рту.
Проверенным и действенным средством считается Галоперидол. Лечение начинается с низкой дозы, которая постепенно увеличивается до наступления эффекта или появления побочных действий.
Бывают эффективны Карбамазепин или Клоназепам.
Показано семейное консультирование и психотерапия.
Синдром беспокойных ног
Это распространенное хроническое заболевание нередко семейного характера с аутосомно-доминантным механизмом наследования. Для данной патологии характерна постоянная потребность шевелить ногами из-за плохо поддающихся описанию неприятных ощущений в глубоких тканях нижних конечностей.
Иногда ощущения распространяются и на руки.
Обычно расстройство проявляется в состоянии покоя, поэтому способно нарушать засыпание. Движения могут продолжаться и состоянии сна (фиксируется при сонографии).
Этиология не установлена.
Синдром беспокойных ног нередко возникает во время беременности, при уремической или диабетической нейропатиях, первичном амилоидозе и злокачественных новообразованиях.
Объективное обследование выявляет основное заболевание или легкую нейропатию, но в большинстве случаев нарушений не обнаруживается.
В случае сочетания синдрома беспокойных ног с железодефицитной анемией нередко лечение анемии приводят к выздоровлению.
Бывают эффективны дофаминергические средства (леводопы, бромкриптин), бензодиазепины (Диазепам, Клоназепам), наркотические анальгетики (Кодеин, Оксикодон).
Фото
 (Пока оценок нет)
(Пока оценок нет)